Regulatory, Ethics, MTA Submission
By: INA-RESPOND CRSS
Introduction
In Indonesia, the conduct of clinical trials in compliance with Good Clinical Practice (GCP) is regulated and overseen by several authorities. These include the Institutional Review Board (IRB)/Independent Ethics Committee (IEC), which safeguards the rights and well-being of trial participants; the National Agency of Drug and Food Control/ Badan Pengawas Obat dan Makanan (BPOM), which reviews and authorizes clinical trials involving investigational products; and the Ministry of Health (MoH), which provides general health trial policy and monitors the implementation of health re-search in the country. In accordance with MoH regulations, all clinical trials conducted in Indonesia must be registered in an official clinical trial registry. Additionally, the MoH oversees the review and approval of Material Transfer Agreements (MTA) for the transfer of biological specimens.
These regulations frameworks play a critical role in ensuring the ethical and scientific integrity of clinical research, but they can also significantly impact the feasibility and timeline of a clinical trial. Therefore, a thorough understanding of each authority’s requirements is essential prior to clinical trial initiation to ensure timely submission and approval.
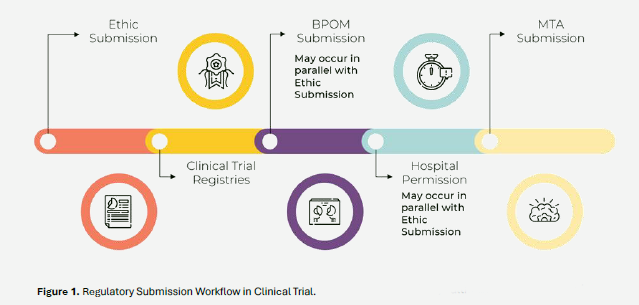
Details of the submission process for obtaining approvals from each regulatory authority were presented during the second session of the training program, held on March 17, 2025. The session covered the clinical trial submission workflow, including Ethics submission, Clinical Trial Registries, BPOM submission, Hospital permission and MTA submission.
IRB/IEC Submission
To initiate a clinical trial, obtaining approval from the IRB/IEC is a critical first step. A complete and well-prepared submission package must be compiled, although specific requirements may vary depending on the policies of each the IRB/IEC. In general, the submission package typically includes:
- Cover Letter
- IRB/IEC Application Form
- Final Protocol
- Protocol Signature Page
- Final Site-Specific Informed Consent Form (ICF)
- Final Case Report Form (CRF)
- Current Principal Investigator’s (PI) Curriculum Vitae (CV) & valid Good Clinical Practice (GCP) certificate
- Supporting documents, such as Insurance Certificate, Investigator’s Brochure (IB), Hospital Accreditation Certificate, Lab Accreditation Certificate & other documents as required by the IRB/IEC.
INA-RESPOND has the option to designate a Central IRB/IEC to provide ethical approval for studies conducted across multiple sites within the network. When this centralized review process is utilized, an IRB Reliance Agreement must be established between the involved institutions and the designated Central IRB/IEC to formalize the arrangement. This agreement ensures that the designated IRB of Record has clearly defined responsibilities and provides consistent ethical oversight across all participating sites. However, if a participating site is still required to obtain approval from its Local IRB/IEC, the site retains full responsibility for submitting the application and managing all necessary reporting and correspondence with the local IRB/IEC. The timeline for review may vary depending (1-2 months) on the policies and processes of each IRB/IEC institution, which must be considered during trial planning. Careful planning and understanding of the IRB/IEC submission pathways — whether centralized or local — are essential for avoiding regulatory delays and ensuring the trial is ready to proceed as scheduled.
Clinical Trial Registries
In accordance with the MoH regulations, all clinical studies conducted in Indonesia must be registered in the national clinical trial registry at https://ina-crr.kemkes.go.id/id, coordinated by Indonesia Clinical Research Center (INA-CRC). Registration should be completed after obtaining ethical approval from the IRB/IEC and Clinical Trial Approval from BPOM.
ICH E6(R3) under principle number 9 mentioned that timely registration on publicly accessible and recognised databases and public posting of clinical trial results, so all clinical trials should be registered in the ClinicalTrials.gov. This step ensures that the study is officially documented and transparent before it begins.
The registered information must include any details about the study, such as the trial title, objectives, methodology, study duration, participating sites, and investigators. Keeping this information accurate and up to date is important throughout the study period. Any significant changes to the study, such as protocol amendments, site updates, or changes in the investigator team, must also be promptly reflected in the registry. Maintaining accurate registration records not only fulfils regulatory obligations but also supports transparency, promotes scientific integrity, and build public trust in the research process.

BPOM Submission
A Clinical Trial Approval (CTA) from BPOM is mandatory for all interventional clinical trials before the study is conducted. The submission must be made through a formal application, including a cover letter addressed to the Director of Drug Registration at BPOM.
The document package for CTA submission includes:
- Cover Letter
- BPOM Application Form
- Final Protocol
- Protocol Signature Page
- Final Site-Specific ICF
- IB
- Current PI’s CV and valid GCP Certificate
- Hospital Accreditation Certificate
- Laboratory Accreditation Certificate
- Insurance Certificate (if applicable)
- IRB/IEC Approval Letter and IRB/IEC Attendance List
- Clinical Trial Application Document Check-list
CTA submissions are made through the SIAP-UK online platform at https://siap-uk.pom.go.id/ as mentioned in the Figure 3. It is important to note that BPOM will not issue approval until IRB/IEC approval has been obtained. The timeline for BPOM to review a complete Clinical Trial Application is 20 working days after all required documents have been submitted. The CTA is valid for two years from the date of issuance; however, for ongoing studies expected to be completed in less than two years, BPOM will issue a CTA with a validity period that aligns with the study’s projected completion timeline.

Import License
Following the issuance of the CTA from BPOM, the next regulatory step is obtaining Import License from BPOM which is mandatory for importing investigational products (IPs) used in clinical trial. The documents typically needed for this process include:
- Cover Letter
- Drug Certificate of Analysis (CoA)
- Drug Good Manufacturing Practice (GMP) Certificate
- Drug Importer Name & Address
- Drug Manufacturer Name & Address
- Drug Calculation, Batch Number, Expiry Date
- Summary of Batch Protocol from 3 Consecutive Batches (for biologic products only) to ensure product consistency
- Lot Release (for vaccines)
- Package Insert or IB
- IP Label
- Investigational Product Usage Report (for subsequent applications)
- Clinical Trial Approval (for subsequent applications)
- Document Checklist for Clinical Trial Application (to be filled in the Import License section)
Once the documents are prepared, they can be submitted via the BPOM e-Submission portal at https://e-bpom.pom.go.id/. Approximately, the import license will be released 10 working days after the application documents have been completely submitted. The import license approval is typically valid for one shipment only. For multiple shipments throughout the study, separate applications must be submitted accordingly. To avoid delays in study initiation, it is advisable to prepare and submit the import license application as early as possible after receiving CTA approval.

MTA Submission
An MTA is required for clinical trials involving the shipment of specimens to laboratory located outside Indonesia. The MTA serves as a formal legal agreement between the sending institutions (e.g., study site or the reference laboratory) and the receiving institution (e.g., central lab overseas), and must be approved by the MoH.
The MTA can be prepared either before the study begins or during or end the trial, depending on the agreement with the and it must be fully approved prior to the shipment of any specimens to the Central Laboratory abroad.
The document required for MTA submission:
- Cover letter addressed according to MTA requirement
- Form A (List of Specimen which will be sent and reason)
- Material Transfer Agreement
- List of materials that will be shipped
- Data Sharing Agreement
- Biosecurity and Biosafety Compliance Certification for Material Management
- CV and GCP of the PIs from all sites in Indonesia and outside of Indonesia
- Study Protocol that approved by Ethic Committee
- MoU or Clinical Trial Agreement between institutions
- IRB/IEC approval from all sites if multicenter study
- For multicenter study, list of institutions and PIs
- BPOM Approval (if needed, example: Clinical Trial specimen)
- Implementing Agreement, including extension agreement (if needed)
- Declaration of competence from the laboratory specimen recipient
- MTA application checklist
- Other documents as needed such as the organizational structure, a permission letter for laboratory use, and laboratory company profiles (if specimens are received by more than one laboratory).
The MTA documents must be submitted through the official MTA Secretariat website at https://mta.kemkes.go.id/ as mentioned in the Figure 5. Prior to the MTA review meeting, the PI should prepare a presentation outlining the study and specimen-related details. It is important to coordinate closely with the PI in advance to finalize the presentation and ensure readiness for discussion.
The PI or designated representative must attend the MTA Review Meeting when invited by the MTA Committee. Participation in this meeting is mandatory to facilitate a clear understanding of the study context and address any committee questions. Once the MTA is approved and specimens are shipped and received by the designated overseas laboratory, the study team is expected to provide the MTA reviewer with a trial progress update as part of ongoing compliance and transparency. The MTA document review process is categorized by risk level, with corresponding timelines: exempted or minimal risk reviews are completed within 6–11 working days, expedited or moderate risk reviews take 9–24 working days, and full board or high-risk reviews require 16–26 working days.
Hospital Permission Submission
Obtaining trial approval from the site hospital director is a crucial step to ensure that trial procedures are carried out efficiently and in alignment with the hospital’s policies and protocols. As part of this process, the trial team must submit a permission letter addressed to the hospital director and comply with all site-specific requirements for conducting the trial. The timeline for hospital permission approval may vary depending on the institution’s internal review process, with some approvals taking up to six months. Therefore, early engagement with the hospital’s clinical research unit or administration is highly recommended. Once approved, hospital permission is generally valid for one year from the issuance date, so renewal approval is mandatory annually.
Typically, this process also includes the negotiation and signing of a Clinical Trial Agreement between the sponsor and the hospital, detailing responsibilities, budgeting, indemnity, and resource provisions. Once all necessary conditions are fulfilled, the hospital usually issues a study team designation letter, formally acknowledging the trial team and authorizing the study’s implementation at the site.
Conclusion
The duration of regulatory submissions varies based on the trial’s type and complexity, making timeline flexibility essential. Each regulatory sub-mission, such as IRB/IEC, BPOM, INA-CRC, MTA Committee, and the hospital permission follows its own process and timeline. Delays in approvals can affect the overall study schedule, so the trial startup team must remain focused, motivated, and attentive to document requirements. Close coordination with sponsors and team members is also critical, especially when responding to regulatory feedback or revision requests. These efforts help keep the trial on track and aligned with the approved protocol and regulatory standards.
Most Commented