
METAGENOMIC NGS (mNGS) TO DETERMINE THE ETIOLOGY OF INFECTION
By: Ungke Anton Jaya
In March 2022, cases of severe acute hepatitis of unknown etiology occurred in many countries beginning in the UK and reported in other countries, including Indonesia. Interestingly, those cases turned out negative after a series of testing to diagnose hepatitis A-E and other common pathogens related to hepatitis infection. In some cases, the preliminary test gave a positive result for SARSCoV2, HHV-6, HHV-7, and Adenovirus. Due to the uncertain etiology of the outbreak, an untargeted metagenomics attempt was made, resulting in the most abundant reads of Adeno-associated virus2 (AAV2). The findings were confirmed by PCR-specific assay. (1) This was an excellent example of metagenomics sequencing as an important tool to identify the etiology of infection. Here we will explore further the potential of this method.
Metagenomics Next Generation Sequencing (mNGS)
DNA sequencing is the method to identify the order of nucleotide sequence of an organism’s genome. This sequence is unique for each organism and can be used to identify a particular organism. If we can identify a piece of DNA fragment sequence and compare it to the GenBank DNA database, we can identify the organism to which the DNA fragments belong. This method can be developed as a tool to diagnose the etiology of infection.
Shotgun sequencing is a laboratory technique for determining the DNA sequence of an organism’s genome. The method involves randomly breaking up the genome into small DNA fragments that are sequenced individually. Later, a computer program looks for an overlapping sequence in the DNA fragments, using them to reassemble the fragments in their correct order to reconstitute the genome. (picture 1)
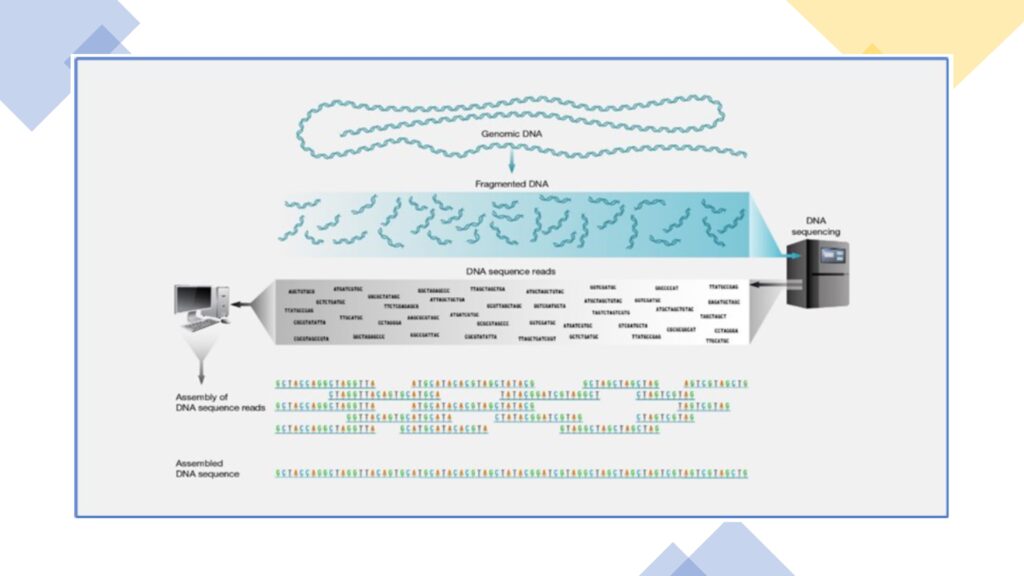

Primer is not always required in sequencing with the shotgun method, in contrast with conventional Sanger sequencing which primer is always needed. This shotgun approach is the basic principle for metagenomics sequencing.
Next Generation Sequencing (NGS) technology is the second generation of sequencing after Sanger sequencing. NGS generates massive, ultra-high throughput sequences and can be done without pre-knowledge of the sequence target or unbiased sequencing. NGS technology can sequence in parallel any DNA template present in a sample. The platform becomes a revolutionary breakthrough that excites scientists to identify what organisms live in samples as we can identify the organisms simply by detecting the presence of their DNA without culturing. Metagenomics NGS (mNGS) is the study of the structure and function of genome sequences analyzed from all the organism’s microbe, i.e., bacteria, viruses, parasites, in a bulk sample such as on human skin, in the soil, or in water. mNGS platform opens the possibility to reveal 98% of the previously unstudied and uncultured microbiome in the environmental sample, drastically adding new information. The sequence data generated from metagenomics NGS is massive and conducted without the need for prior knowledge of a specific pathogen, which is why it is known as unbiased or agnostic high throughput sequencing.
mNGS is a powerful technique that enables the detection of the full spectrum of pathogens present in any specimen in a single test. mNGS can also provide information on the relative abundance of the organism in the sample containing a community of organisms such as the human intestine or skin. The relative abundance can lead to drawing conclusions on the pathogen that may cause the disease.
In contrast to metagenomics NGS, the conventional Sanger sequencing requires a primer to start the sequencing process and generates a sequence of 500 to 1000 nucleotide bases length in one direction, forward or reverse only (Picture 2). To sequence a long fragment of a genome, many primers are pre-designed using a reference sequence of the target organism. Sanger sequencing requires both forward and reverse primers to obtain complete sequencing of the targeted genome. Sanger sequencing requires a DNA template that is homogeneous and abundant from a targeted organism that is usually generated through PCR amplification. If DNA templates contain DNA mixtures from many organisms, the Sanger sequencing reaction will generate an unspecific sequence peak that is interpretable. The condition that makes metagenomics sequencing using the Sanger method will be extremely difficult. Sanger sequencing is more suitable to sequence targeted specific pathogens from PCR products.
Clinical metagenomics NGS (mNGS) for pathogen identification
The metagenomics NGS platform has strong advantage and power to be used as a diagnostic tool for the etiology of infection, including bacterial, fungal, and viral. In reality, it remains a very challenging process until now. The clinical sample is over-whelmed by human cells with a genome size of 3.200 Mb (Megabase) per cell that outnumber compared to microbes, viruses (4 kb to 1.3 Mb), bacterial (0.6 to 8.0 Mb), or fungal (8.97 – 177.57 Mb). The throat swab specimen always contains both human epithelial cells and commensal bacteria that make detection of the viral genome as potential etiology like finding a “needle-in-a-haystack” situation. The effort will be less complicated if clinical samples come from sterile body sites such as serum and cerebrospinal fluid, which normally contain no pathogens. In the attempt to detect the viral genome, following nucleic acid extraction, human genomic can further be depleted by enzymatic reaction, leaving the viral RNA more exposed for detection.
Application of metagenomics NGS for diagnosis of bacterial infection can be conducted by targeted sequencing using PCR amplification targeting universal bacterial 16s ribosomal RNA (rRNA) gene to increase sensitivity and specificity. This method is not unbiased sequencing but a targeted metagenomics NGS. It is very common that mNGS output normally detects multiple bacterial genome. To determine the etiology of infection, complex bioinformatics analysis and algorithm will be followed, such as whether the bacterial is known as a human pathogen or not and the relative abundance of certain bacterial genomes reflected as the abundance of sequence read/fragment. A similar approach can be used to identify suspects of fungal infection using universal PCR amplification targeting 18s rRNA and the internal transcribed spacer (ITS) gene.
The more challenging process is identifying viral infection with unknown etiology since virus does not have a universal sequence. The viral metagenomics approach can be made by PCR amplification using multiplex primer followed by an mNGS assay. For example, mNGS used 285 and 256 primer pairs to identify 46 virus species causing hemorrhagic fevers. (3) or sequencing whole genome virus SARSCoV2 using ARCTIC primer set from clinical respiratory sample. Another approach is using random primer PCR approach that was successfully used to identify viruses in samples from Sepsis cases with unknown etiology. (4) Another viral metagenomics approach is using capture probe enrichment VirCap Seq-VERT that employs ~2 million probes covering the genomes of members of the 207 viral taxa known to infect vertebrates, including humans. The probe set has been commercially available. (5) All approaches apply untargeted or unbiased high throughput sequencing.
Development of metagenomics NGS for detection of infection improves very fast along with the growing capacity to do NGS and the decrease in the cost of NGS reagents and consumables. Metagenomics NGS still faces major challenges to becoming a reliable diagnostic tool, with main challenges like assay validation, reproducibility, high cost, long turnaround time, and obtaining regulatory approval. In fact, several groups have successfully validated mNGS in Clinical Laboratory Improvement Amendments (CLIA)-certified clinical laboratories for diagnosing infections, including meningitis or encephalitis, sepsis, and pneumonia, and these assays are now available for clinical reference testing of patients.
In Indonesia, metagenomics NGS for bacteria has been initiated in researching the human gut microbiome and antibiotic resistance gene of Mycobacterium tuberculosis. Recently NGS has been used in massive efforts for targeted sequencing of genomic surveillance of SARSCoV2. The activity has provided the country with the infrastructure capacity, instrument, reagent, bioinformatics skills, and experience in NGS application. The NGS capacity has the potential to switch to metagenomics NGS if needed to identify the diagnosis of infection, such as in the diagnosis of acute hepatitis cases with unknown etiology in Indonesia.
Clinical metagenomics NGS application for identification of infection still faces many challenges in becoming a diagnostic tool for patient treatment. However, referring to the promising potency, the development of mNGS is currently on the fast track to becoming a powerful tool for diagnosing infection.
Reference:
- Morfopoulou S, et al. GENOMIC INVESTIGATIONS OF ACUTE HEPATITIS OF UNKNOWN AETIOLOGY IN CHIL-DREN. medRxiv [Internet]. 2022 Jan 1;2022.07.28.22277963. Available from: http://medrxiv.org/content/early/2022/07/28/2022.07.28.22277963.abstract
- Eric G. Shotgun sequencing definition. National Human Genome Research Institute. (https://www.genome.gov/genetics-glossary/Shotgun-Sequencing).
- Brinkmann A, et al. Development and preliminary evaluation of a multiplexed amplification and next generation sequencing method for viral hemorrhagic fever diagnostics. PLoS neglected tropical diseases. 2017 Nov;11(11): e0006075.
- Anh NT, et al. Viruses in Vietnamese Patients Presenting with Community-Acquired Sepsis of Unknown Cause. Journal of clinical microbiology. 2019 Sep;57(9).
- Thomas B, et al. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. mBio [Internet]. 2015 Sep 22;6(5): e01491-15. Available from: https://doi.org/10.1128/mBio.01491-15.
Most Commented